Lung tissue samples¶
To get started and become familiar with the types of analyses you can perform using SpatialRNA, we recommend going through the first three tutorials before scaling up your analysis to larger sets of samples.
In this case study, we analyse 45 Xenium lung tissue samples from GSE250346, including both healthy and fibrotic conditions.
A total of 343 genes were measured across samples, each approximately 3–5 mm in diameter, yielding around 299 million detected transcripts Vannan, Lyu et al, 2025.
We integrate transcripts from all 45 samples by training a GNN model on the combined subgraphs. Graphs and subgraphs are first constructed for each sample individually, and then joined for integrative analysis.
Preparing input transcripts¶
Each sample’s input transcripts should be stored in a CSV file (SampleName.csv) containing both spatial coordinates and gene names.
Before use, all transcript lists must be filtered to remove:
Control probes
Low-quality detections (e.g.,
qv < 20)
For multiple samples, the input CSV files should follow the directory structure:
data_dir_name/sampleName1/raw/sampleName1.csv
data_dir_name/sampleName2/raw/sampleName2.csv
data_dir_name/sampleName3/raw/sampleName3.csvProcessing steps¶
After the input transcripts are prepared as above, we can run spatialRNA to construct spatial transcript radius graphs for each sample, and we sample a subgraph from each sample which will be used for training the GNN model.
Now data dir will have new foler “processed” which stores the graph objects, and subgraph data objects
data_dir_name/sampleName1/processed/sampleName1_data_tile0.pt
data_dir_name/sampleName2/processed/sampleName2_data_tile0.pt
data_dir_name/sampleName3/processed/sampleName3_data_tile0.ptdata_dir_name/sampleName1/subgraph/sampleName1_data_tile0.pt
data_dir_name/sampleName2/subgraph/sampleName2_data_tile0.pt
data_dir_name/sampleName3/subgraph/sampleName3_data_tile0.ptIn this step, we train the GNN model on the combined subgraphs from all 45 samples, as defined in the Snakemake workflow.
All 45 subgraphs are first loaded and merged into a joined graph.
Mini-batches of training graphs are then loaded onto the GPU for efficient training (see
./code/run_GNN_training.py).
If your machine does not have sufficient resources to load all subgraphs into memory, you can use the
SpatialRNAOnDiskDatasetclass to manage on-disk batched loading and reduce CPU memory requirements. For example:myod = SpatialRNAOnDiskDataset(root="../data/", pt_dir="subgraph")For a more compete example with the SpatialRNAOnDiskDataset usage, please refer to the Case Study of the 5K Ovarian Cancer data.
In this final step, we compute the latent embeddings of all transcripts in each sample.
To identify transcript-based molecular niches, we perform clustering on the combined embeddings matrix.In this case study, we applied a Gaussian Mixture Model (GMM) to cluster transcripts into 12 niches, using the Pycave library, which provides GPU acceleration for clustering.
We then use either the
plot_pixelorplot_hexfunction to visualize the molecular niches spatially.
Below, we display pixel plots for several representative samples:
A healthy lung tissue¶
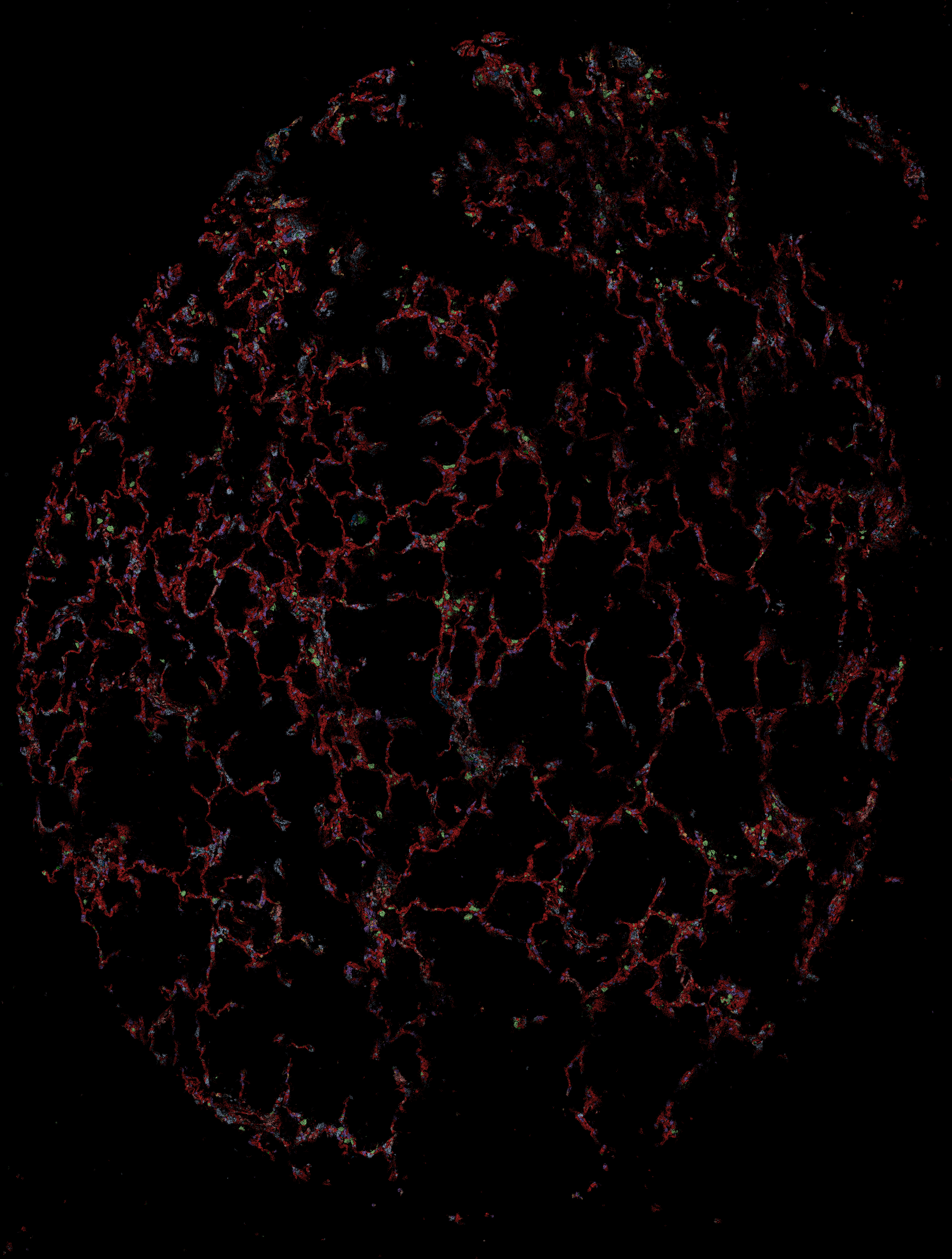
A fibrotic lung tissue¶

- Vannan, A., Lyu, R., Williams, A. L., Negretti, N. M., Mee, E. D., Hirsh, J., Hirsh, S., Hadad, N., Nichols, D. S., Calvi, C. L., Taylor, C. J., Polosukhin, Vasiliy. V., Serezani, A. P. M., McCall, A. S., Gokey, J. J., Shim, H., Ware, L. B., Bacchetta, M. J., Shaver, C. M., … Banovich, N. E. (2025). Spatial transcriptomics identifies molecular niche dysregulation associated with distal lung remodeling in pulmonary fibrosis. Nature Genetics, 57(3), 647–658. 10.1038/s41588-025-02080-x